

Inherited neuromuscular disorders include muscular dystrophies, a group of genetic conditions characterized by progressive skeletal muscle involvement. These disorders arise from mutations that affect muscle-structure or repair proteins, including dystrophin. Altered or absent dystrophin compromises fiber stability and contributes to muscle weakness and reduced endurance.

When dystrophin or related proteins are insufficient, muscles do not maintain typical function. Descriptions commonly include difficulties with walking or swallowing, depending on subtype and stage. Presentations occur across the lifespan, with many forms identified in childhood. X-linked dystrophinopathies are more frequently reported in males.
The underlying cause is a pathogenic variant in genes that support muscle integrity. Protective functions against contraction-related injury are reduced when such genes are altered or absent. Each recognized type corresponds to specific genetic changes; inheritance patterns vary and include X-linked, autosomal dominant, and autosomal recessive transmission.

Variants may be inherited or occur de novo in parental germ cells. Duchenne muscular dystrophy (DMD) is among the most prevalent dystrophinopathies with early childhood onset, whereas Becker muscular dystrophy (BMD) typically presents later, often in adolescence or adulthood. Age at presentation and rate of progression differ between these conditions.

BMD generally progresses more slowly than DMD. Early features described in summaries include muscle cramps, stiffness, proximal limb weakness, and a waddling gait. Some individuals, particularly with dystrophinopathies, may have learning or attention difficulties; reported severity varies and is influenced by specific genetic alterations.
Distinct muscular dystrophies are defined by characteristic patterns. Myotonic dystrophy (Steinert disease, DM1) features delayed muscle relaxation (myotonia) and frequently involves facial and cervical muscles. Facioscapulohumeral muscular dystrophy (FSHD) is another well-characterized form with a different genetic basis and distribution of weakness.

FSHD often begins with facial and shoulder-girdle muscles. Congenital muscular dystrophies are identified in infancy or before two years of age. Limb-girdle muscular dystrophies primarily involve hip and shoulder musculature. Rates of progression span a broad range across and within subtypes.
Potential consequences described in clinical sources include progressive mobility limitations. Many individuals transition to assisted ambulation or wheelchair use over time. Muscle shortening (contractures) can develop and contribute to reduced joint range of motion.
Contractures, respiratory muscle involvement, and spinal curvature (scoliosis) are reported complications in several forms. Cardiac manifestations, including cardiomyopathy or conduction abnormalities, may occur in specific subtypes. Oropharyngeal muscle weakness can be associated with difficulties in swallowing.
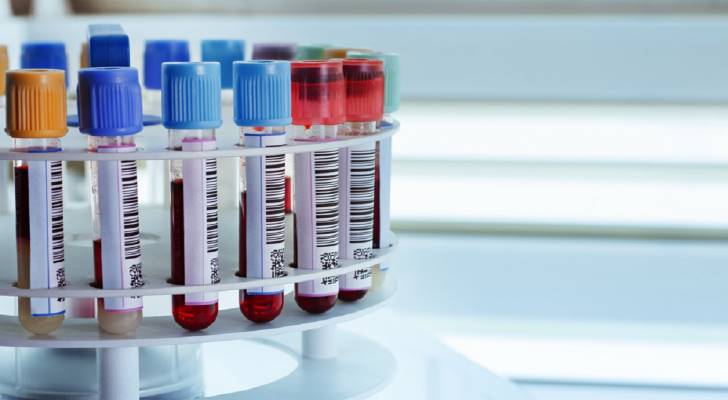
Assessment typically integrates laboratory and physiologic studies. Serum enzymes such as creatine kinase are often elevated in active muscle injury. Molecular genetic testing can identify causative variants. Electromyography evaluates muscle electrical activity, and muscle biopsy may be used to examine fiber structure and protein expression when indicated.
Learn more about muscular dystrophy at National Institute of Neurological Disorders and Stroke (NINDS).